| Characteristics: |
| Correspondence Analysis |
| Planar embedding |
| Interpretation |
| Storing annotations without freetext |
| Why? |
| How? |
| To what end? |
Preprocessing of hybridization intensities
Normalization
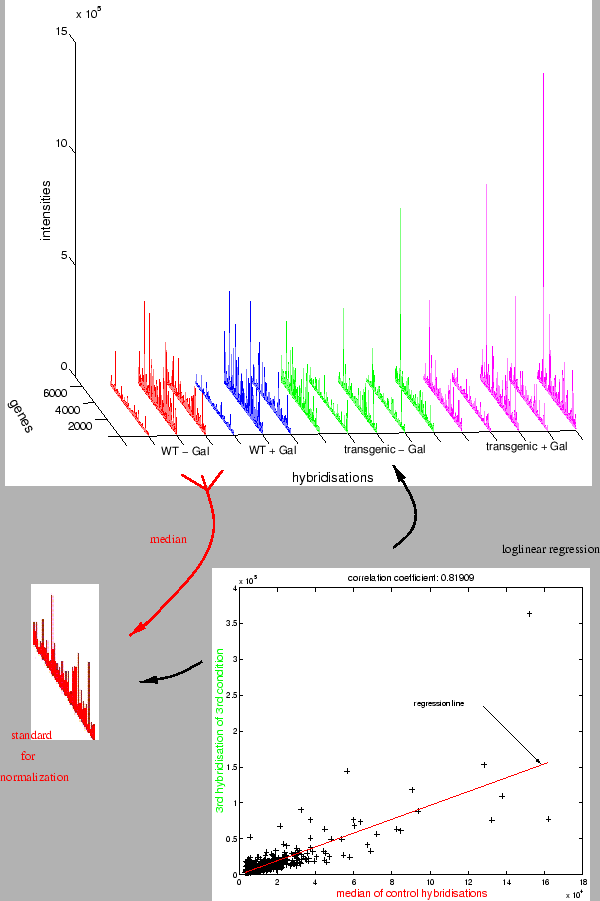
Prior to high-level analysis, data have to be normalized and filtered. In M-CHIPS, preprocessing starts with normalization of raw signal intensities. Different levels of background may result in additive offsets, or different amounts of mRNA or different label incorporation rates may lead to multiplicative distortions among the hybridizations. The normalization is based on robust affine-linear regression, i.e. it corrects for additive offsets and multiplicative distortions at the same time. The algorithms fit one measurement versus a control measurement. The performance may be judged from the scatterplot of the raw data (measurement versus control measurement, Fig. 1). In this plot, a regression line represents the multiplicative distortion (slope) and additive offset (intersect) determined by the fitting algorithm. The performance of the fit is visible in how well the regression line matches the central dense part of the cloud. Furthermore it can be observed which properties of the raw data led to an eventually suboptimal result. The scale of the plot can be switched between linear and double-logarithmic. In log scale, the regression line appears as a curve whose curvature depends on the additive offset between the two measurements.
M-CHIPS implements two algorithms as described in [1] and [2]. From amongst the options given in [1] I use the 5% quantile1 of each hybridization as the additive offset to subtract initially. The original algorithm results in a shift of the original data to a lower intensity level, making it necessary to ignore values below a certain threshold. For correspondence analysis it is advisable to use another normalization method because low intensity signals have to be kept in order to avoid missing data. Instead, I shift all hybridizations additively to a higher range, in order to prevent overly biasing CA by the large relative error common to low intensities. This shifting is done such that the 5% quantiles coincide with that of the control measurement. For both algorithms, the set of trusted spots of unvaried expression taken into account for fitting can be specified. In most cases, the share of differentially transcribed genes is low, enabling to use the entire set for normalization. Otherwise, external control spots reporting defined mRNA concentrations or trusted housekeeping genes have to be used. For the former, defined amounts of complementary mRNA are added to the samples prior to the labelling step. For the latter, a text file is imported into M-CHIPS, listing genes that are trusted to be constitutively transcribed under the investigated experimental conditions.
In order to normalize a whole multiconditional experiment, the above
algorithms are iterated. All measurements are iteratively normalized
with respect to one and the same control condition, such that they
can be compared afterwards. M-CHIPS discriminates between mono- and
multichannel experiments, applying different control measurements
and iteration steps. For monochannel (e.g. radioactive) data, each
measurement is normalized versus the genewise median of the hybridizations
for the control condition, resulting in absolute intensities (Fig.
1).
|
For multichannel hybridizations, the channel belonging to the control condition serves to normalize the other channel(s) of the same hybridization. Here, the normalized intensity values are not analyzed as such, but result in intensity ratios, calculated immediately after normalization. Normalization requires, that each hybridization comprises one channel obtained from the same control condition.
Filtering
Prior to high-level analysis, M-CHIPS provides a means to select genes which fulfill the following criteria: considerable absolute expression level in at least one of the conditions; substantial change relative to the control condition in at least one of the other conditions; and reproducibility in the separation from the control condition (Fig. 2) in at least one of the other conditions.
Intensity. For many arrays and experiments, the majority of genes spotted on the array are not expressed to a measurable amount. While displaying notable ratios due to measurement fluctuations, they can be eliminated by means of an intensity filter. For monochannel experiments, meaningful intensity levels are obtained by the normalization procedure. For more than one channel, apart from reflecting a low concentration of the corresponding mRNA, a low signal can be caused e.g. by high concentrations of differently labeled mRNA taking the majority of the binding sites of the spot. Therefore, multichannel intensity values are not valid as such but only in conjunction with the other channel(s) of the same hybridization. This establishes the above requirement of one and the same control condition on each hybridization for comparability. For the same reason, normalized multichannel intensities cannot be used for high-level analysis nor for intensity filtering. However, they can serve to compute ratios reflecting the relative abundance of a certain mRNA sequence under a specific condition compared to a control condition. To compute intensity levels from multichannel ratios for filtering purposes, these ratios are multiplied with an average control measurement, being the gene-wise median of the absolute intensities of the control channels. This average is a more stable basis for the determination of intensity levels. Apart from eliminating outliers by averaging repeated measurements, this procedure accounts for the above example of highly abundant differently labeled mRNA. Provided that less than 50% of the non-control conditions under study show such a high abundance of a specific mRNA, the intensity level of the control condition for that mRNA will not be low due to lack of binding sites.
Ratio. For multichannel data, ratios for each measurement are computed by dividing each normalized non-control channel gene-wise by the control channel of the same hybridization. For monochannel data, each hybridization is divided by the gene-wise median of all control hybridizations.
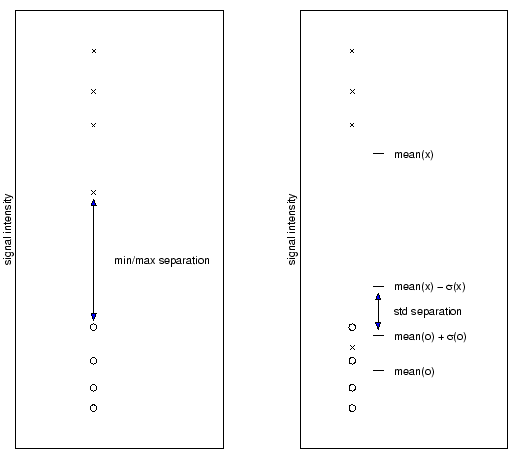
Separation. Apart from intensity and ratio filters, reproducibility
measures [1] are applied to extract genes that are
reproducibly up- or down-regulated. These measures integrate repeatedly
performed measurements for the same experimental condition by providing
the separation from a control condition (Fig. 2).
|
Apart from being filter criteria for the set of genes, they are plotted versus the average intensity level and ratio as a measure for quality control. Moreover, they have been successfully applied directly as high-level analysis input (not shown). For this, all negative separation values are set to zero and attached with a positive sign for upregulation or a negative one for downregulation, instead. Thus they can be viewed as log ratio signal, which is suppressed by imperfect reproducibility.
Filtering. To filter out genes displaying intensities clearly above the detection limit, significant relative change and good reproducibility of this change, intensity-, ratio- and separation-thresholds can be applied. Genes not satisfying these constraints are discarded. One can also discard genes above rather than below a threshold which proved to be useful to account for saturation effects occuring e.g. if radioactively labeled arrays were exposed too long. In general, M-CHIPS provides AND-combination of three independent constraints, each of which can be defined as
- selecting genes above or below a certain threshold;
- that threshold applies to raw or normalized intensities or ratios, condition-medians of normalized intensities, ratios of condition-medians of intensities, min-max separations, or standard-deviation separations;
- it may be operational only within a specified set of conditions, e.g. all the conditions under study.
Bibliography
- 1
-
T. Beißbarth, K. Fellenberg, B. Brors, R. Arribas-Prat, J. M. Boer, N. C.
Hauser, M. Scheideler, J. D. Hoheisel, G. Schütz, A. Poustka, and
M. Vingron.
Processing and quality control of DNA array hybridization data.
Bioinformatics, 16:1014-1022, 2000. - 2
-
K. Fellenberg, N. C. Hauser, B. Brors, A. Neutzner, J. D. Hoheisel, and
M. Vingron.
Correspondence analysis applied to microarray data.
Proc. Natl. Acad. Sci. U.S.A., 98:10781-10786, 2001.
Footnotes
- ... quantile1
- Ranking the genes by signal intensity for a given hybridization, the maximum intensity of the lowest 5% of the genes is referred to as 5% quantile of the hybridization.